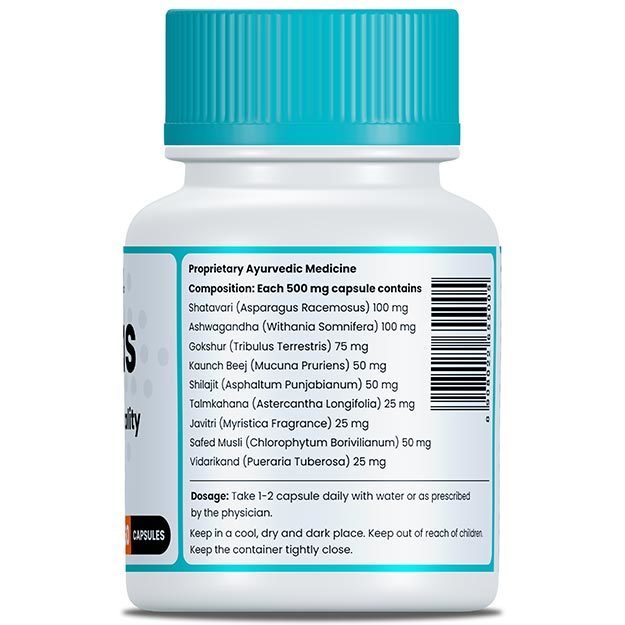
There is no cure for CPT2 deficiency. The myopathic form is a lifelong condition, and management of the disease requires multi-directional approaches - your doctor may recommend medications, dietary supplements and diet or lifestyle modification. The healthcare provider might also refer you to consult a dietician to keep a check on your diet.
Eating at regular intervals: The best way to prevent symptoms from recurring is to keep eating at regular intervals. Parents are advised to pay attention to the eating patterns of infants and young children with CPT2 deficiency - young patients should be fed frequently to prevent a metabolic crisis. Usually, infants should eat every four to six hours. It is critically important to make the child eat even during the night. Waking up and eating once or twice during the night prevents episodes of metabolic crisis. Your doctor and dietician will give you an appropriate diet plan for your child. A slightly modified diet plan is also given to the parents or patients and should be followed during active illness.
Diet: CPT2 deficiency is a fat metabolism disorder. As such diet is extremely important in order to avoid disease symptoms. Usually a low fat, high carbohydrate food plan is recommended. Carbohydrates provide several types of sugars to the body and can be used as a source of energy. Sometimes, eating sugar during an episodic attack of symptoms can provide relief.
Patients with CPT2 deficiency can’t consume long-chain fatty acids - these include olive oil, fish, meat and nuts. Most of the fat in their recommended diet consists of medium-chain fatty acids - these include dairy products and coconut oil.
Long-chain fatty acids usually have 13-21 carbon atoms where are medium-chain fatty acids have 6-12 carbon atoms and are easier to break down for energy.
MCT oil and Triheptanoin: Medium Chain Triglyceride oil (MCT oil) is often used as part of the food plan for people with CPT2 deficiency. This special oil supplement has medium-chain fatty acids that can be used in small amounts for energy.
Triheptanoin is a triglyceride made up of three seven-carbon fatty acids. (The name triglycerides comes from the fact that each molecule contains tri or three molecules of fatty acids. Fatty acids, in turn, can be of several types. Those with seven carbon atoms fall in the medium-chain category.) Triheptanoin is thought to break down and produce ketone bodies and propionyl-CoA. Clinical trials are on, and researchers are looking into the effectiveness of this supplement.
L-carnitine: L-carnitine is an amino acid is found abundantly in heart muscle and skeletal tissue. It is sometimes prescribed as a supplement. It functions primarily as a shuttle for fatty acids, transporting them across mitochondrial membranes into the mitochondria, where they are used for energy production.
Riboflavin: Riboflavin or vitamin B2 supplements may induce a dramatic improvement of muscle symptoms related to CPT2 deficiency disorder.
Bezafibrate: Bezafibrate is a drug which reduces the lipid level in the body. It is usually prescribed for fatty-acid oxidation disorder. Clinical trials have shown that it can improve muscle function in people living with CPT2 deficiency.
AMP-activated protein kinase (AMPK): Exercise, drugs like metformin, rosiglitazone and hormones such as leptin and adiponectin activate AMPK (an enzyme) in the skeletal muscles and increase fatty-acid oxidation in muscles, thereby providing relief from the symptoms of the disease.



 Download App
Download App